Sedative Alpha-2-adrenergic Agonists
Alpha-2 adrenergic agonists, or α2-adrenomimetics represent a pharmacological group of substances that induce stimulation of α2-receptors of the adrenergic system. In clinical medicine, α2-adrenomimetics are used as sedative agents, in contrast to α1-adrenomimetics, which belong to the group of hypotensive drugs.
Effects of alpha-adrenomimetics on the human body[1,67]
Central nervous system
Cause pronounced sedation. Eliminate anxiety states. Exert central analgesic action. Induce hypothermia.
Cardiovascular system
Characterized by biphasic effects on blood pressure: brief elevation followed by prolonged reduction. Lead to decreased heart rate (bradycardia). Impair coronary and cerebral blood flow.
Respiratory system
Depress the respiratory center only in doses significantly exceeding therapeutic levels. Clonidine may relieve bronchospasm induced by other substances.
Digestive system
Induce nausea and decreased salivary secretions. Block secretion of water and electrolytes into the small intestine lumen and may be effective in treating secretory diarrhea.
Urinary system
Possess diuretic action.
Endocrine system
Stimulate growth hormone secretion. Inhibit insulin production, increase blood glucose levels.
Ophthalmological effects
Cause mydriasis, reduce intraocular pressure.
Advantages and Limitations. Incapacitants from the opioid, inhalational, and dissociative anesthetic groups are characterized by rapid and profound consciousness depression, which can lead to trauma from falls or seizures, asphyxia due to tongue retraction or aspiration of vomitus, and development of positional compression syndrome[2].
Incapacitants from the α2-adrenomimetic group belong to the second generation of non-lethal chemical weapons (EA 5995, EA 5955, EA 5978)[11]. The mechanism of action of α2 - adrenomimetics differs from other sedative and narcotic agents — even in high doses they do not cause anesthesia[21], after medetomidine administration patients lie calmly in bed but are easily roused to full consciousness[2]. Under the influence of adrenomimetics, the subject may retain consciousness but completely loses the ability for coordinated active actions.
Most α2-adrenomimetics have negative effects on the cardiovascular system, which limits their use as standalone incapacitants. In this regard, in recent decades their application in non-lethal weapons has been considered primarily in combination with other CNS-depressing substances: anesthetics, opioids, and tranquilizers.
Adrenergic Agonists as “Knockout Agents”
In 1942, American pharmacologist N. Fabricant proposed a drug under the trade name Privine for treating rhinitis, whose active substance was naphazoline — an adrenomimetic from the imidazoline derivative group[27]. The drug gained widespread clinical use worldwide and is currently known in former Soviet countries under the name Naphthyzin.
After the onset of mass clinical use of Privine, reports began appearing about sedative side effects of the drug in children and individuals with increased sensitivity, but this effect initially did not attract researchers' attention.
In 1952, the Central Intelligence Agency initiated a large-scale research program on human mental activity control under the code name MKULTRA. One of the priority directions of the program was the search for "knockout agents" (K-agents) — chemical substances causing temporary loss of consciousness in humans. Researchers working under contract with the CIA systematically studied substances capable of inducing unconscious states.
In 1952, one of the medical consultants collaborating with the CIA drew attention to a study in which schizophrenia patients were given a drug containing Privine, which "produces a shock reaction which does not involve convulsions"[56]. This effect generated significant interest among intelligence agency researchers. The MK-Ultra program documents preserved the structural formula of a promising compound — "Miracle Privine," representing a hybrid of Privine and Chlorpromazine, one of the strongest CNS depressants known at that time. According to the document, such substances were planned for use in a "special project on the covert use of chemicals against individuals and small groups"[57]. The results of these studies remain unknown, as most MK-ULTRA program documents were destroyed in 1973.
 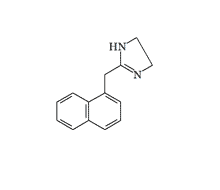
Naphazoline |
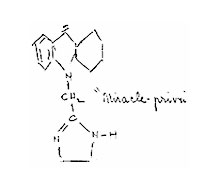 "Miracle Privine" according to the CIA |
 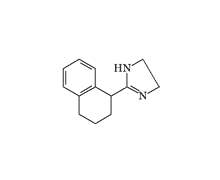
Tetrahydrozoline |
The active component of the ophthalmological preparation "Visine" is tetrahydrozoline, which has structural similarity to Privine but lacks the methylene bridge. In addition to vasoconstrictor action, it exerts pronounced hypnotic effects — a dose of 2–3 mg of the drug induces sleep within 20–30 minutes after administration. In 1959, Pfizer obtained a patent for using tetrahydrozoline in capsulated form as a sleeping aid[36]. However, severe side effects[37], including fatal outcomes[38], led to abandonment of this application.
Clonidine, better known in former Soviet countries under the trade name "Clopheline," was first synthesized in 1962 by the German company Boehringer Ingelheim. The drug was initially planned for use as a nasal decongestant due to its vasoconstrictor properties. In animal experiments, clonidine demonstrated hypotensive action and bradycardia. The sedative properties of Clonidine were discovered by accident when the secretary of research group leader Dr. M. Wolf, who suffered from rhinitis, applied the experimental drug and fell into deep sleep lasting about a day. Subsequent calculations showed that she had received a dose equivalent to 20 Clonidine tablets[28,29].

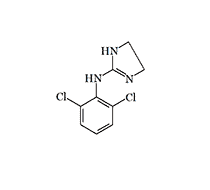
Clonidine (Clopheline, Catapres)
In the mid-1980s in the USSR, medical institutions began receiving masses of patients in unconscious states with arterial hypotension and bradycardia. In patients regaining consciousness, any attempts at verticalization led to syncopal states. Some patients complained of amnesia, auditory and visual hallucinations, color perception disorders — objects were perceived in red-black tones[5]. All cases represented criminal poisonings with Clopheline/Clonidine, typically after consuming alcoholic beverages with unfamiliar persons.
By the mid-1990s, Clopheline poisoning cases had acquired epidemic character. According to E.A. Luzhnikov (1995), acute Clopheline poisonings comprised up to 30% of the total number of patients with drug intoxications[42]. A significant number of victims with mild to moderate poisoning did not seek medical help, and Clopheline was often the cause of sudden deaths outside medical institutions.
Data on Clonidine toxicity are contradictory. Despite the high therapeutic index in laboratory animals reaching several thousand[33], taking 4 mg of Clonidine (20 tablets) combined with ethanol can cause respiratory arrest in humans[34]. At doses above 0.01 mg/kg, clonidine induces hypotension and bradycardia; respiratory depression and apnea may develop at doses from 0.02 mg/kg[32]. Nevertheless, fatal outcomes are relatively rare. According to the American Association of Poison Control Centers (AAPCC), out of 6000 overdose cases in children and adolescents, only one ended in death of a two-year-old child[31].
The possibility of using clonidine as a chemical weapon was discussed immediately after its discovery[40], and its chemical structure determined the direction of incapacitant development from the adrenomimetic group for subsequent decades. Initially, it was believed that loss of combat effectiveness under clonidine exposure was primarily due to pronounced hypotensive action.
Development of Adrenomimetic Incapacitants in USA (1960–1990s)
In 1957, researchers at Pfizer reported results of animal testing of 2-(1-naphthylamino)-2-oxazoline — a representative of a new class of CNS depressants, and announced plans to publish detailed data on its pharmacological properties[35]. However, the publication did not follow, probably due to the decision on military capabilities of drugs in this group.
In 1964, pharmaceutical company Pfizer concluded a contract with Edgewood Chemical Research, Development and Engineering Center (ERDEC) to search for and study drugs capable of causing temporary loss of combat effectiveness — incapacitants. The biological activity of one of the first synthesized compounds under code 400,386 proved to be "commensurate with requirements for modern incapacitants"[19]. By chemical structure, substance 400,386 represented naphazoline with the methylene bridge replaced by an amino group. During further research, drug 400,483 was discovered with higher activity, which at a dose of 0.001–0.003 mg/kg caused ataxia, decreased motor activity, arterial hypotension, and bradycardia in experimental animals. The therapeutic index (LD₅₀/ED₅₀) of these substances reached several thousand[19].
 |
 |
 |
 |
 |
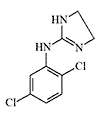 |
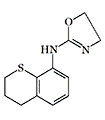
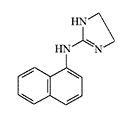
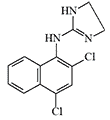
Some candidates for incapacitant chemical agents from Pfizer (on the left) and DuPont (on the right)
Simultaneously, another American chemical company DuPont conducted tests of Tetrahydronaphthyloxazoline (119,902) — the main competitor to substance 400,386. Subsequently, research at DuPont focused on aminooxazoline and aminothiazoline derivatives. Compounds of this group, when inhaled as aerosols, caused temporary immobilization of laboratory animals lasting from 30 minutes to several hours, with the effective dose being 300–750 times smaller than the lethal dose[20].
Despite their attractiveness, the new DuPont incapacitants were inferior in activity to other known incapacitants of that period. Animal immobilization required relatively high substance concentrations — at least 100–500 mg·min/m³, which was several times higher than LSD-25 and BZ indicators[20]. The onset of action was also characterized by relative slowness — animals fell into prostration 3–5 minutes after exposure began. To enhance CNS-depressing action and reduce bradycardia, oxazoline and thiazoline derivatives were tested in combination with cholinergic substances[21], particularly substance 119,902 with deliriants EA 3443 and agent BZ[58].
After Berthelsen and Pettinger's discovery of α₁- and α₂-adrenoreceptor subclasses in 1977, researchers gained the opportunity for selective screening of sedative α₂-adrenomimetics lacking side hypotensive effects.
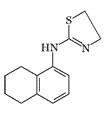
Medetomidine (Domitor®) — a selective α₂-adrenoreceptor agonist, was developed by Finnish pharmaceutical company Orion Corporation in 1987 and was initially used exclusively in veterinary practice. In 1999, the dextrorotatory isomer of medetomidine under the trademark Precedex® was approved for medical use as a sedative agent. The drug is characterized by high activity — administration of 0.12 mg dexmedetomidine to humans induces sleep in 75% of cases.
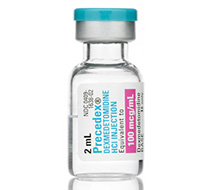
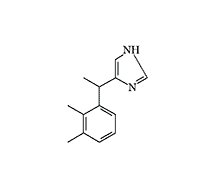
Medetomidine (Precedex)
Unlike opioid analgesics, medetomidine does not depress the respiratory center, but often induces arterial hypotension and bradycardia even at therapeutic dosages.
Medetomidine meets most requirements for chemical incapacitants: it is a pharmacological preparation with well-studied effects on humans, shows activity at low doses, has a high safety index, and a specific antidote.
Due to high lipophilicity, medetomidine has rapid effects on the CNS, and its solutions can be absorbed through the skin. In addition to aerosol form, the use of medetomidine in combination with dimethyl sulfoxide (DMSO) in non-lethal jet or paintball-type weapons is being considered[11,55]. Dimethyl sulfoxide increases skin permeability for adrenomimetics — in one experiment, naphazoline absorption rate increased 25-fold with DMSO addition[41]. When it gets in the eyes, on mucous membranes, or skin, medetomidine may cause irritation.
In the late 1980s — early 1990s, the Edgewood Chemical Research, Development and Engineering Center (ERDEC) continued searching for incapacitants from the selective α₂-adrenomimetic group. Computer modeling was used to study the relationship between chemical structure and sedative action of α₂-adrenomimetics[7]. ERDEC received samples of new drugs in this group from American and European pharmaceutical companies, selecting substances with the highest biological activity for testing. For example, compound A-62033, synthesized by pharmaceutical company Abbott[25,26], proved unsuitable as an incapacitant due to extremely high toxicity — a dose of only 0.0035 mg/kg A-62033 caused life-threatening arterial hypotension in animals[21].
Drug UK-14304 had CNS-depressing effects starting from a dose of 0.001 mg/kg and exceeded medetomidine and naphthylmedetomidine in sedative action[21]. This drug is currently known as a glaucoma treatment under the trademark "Alphagan" (Brimonidine), but structurally similar quinoxaline and quinoline derivatives were synthesized and tested with ERDEC participation in the early 1990s[48].
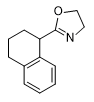 |
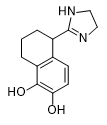 |
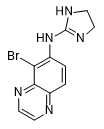 |
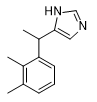 |
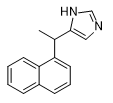 |
| 119,902 | A-62033 | Brimonidine | Medetomidine | Naphthylmedetomidine |
ERDEC was also interested in methylene bridge-substituted[50,59] and 2-imidazoline analogs of medetomidine[49]. Later, the main focus was on studying bicyclic and tricyclic analogs of medetomidine[51,60-62]. The most active compounds obtained were planned to be separated into optical isomers and isolate the more active (S)-stereoisomers[61]. Data on the activity of synthesized adrenomimetics in animal experiments were not published.
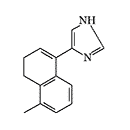 |
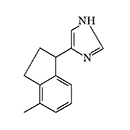 |
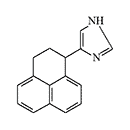 |
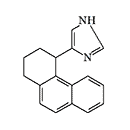 |
Conformationally-restricted analogs of medetomidine synthesized with ERDEC participation in the early 1990s
In 1994, the famous Edgewood Research Center (ERDEC) adopted an ambitious three-year plan to study how adrenomimetics affect humans. The goals were quite specific: to create non-lethal means for suppressing mass riots, protecting embassies, and combating terrorism. Police and the FBI were also extremely interested in obtaining reliable and safe tools for hostage rescue, arresting particularly dangerous criminals, and suppressing prison riots. About 1.3 million dollars was planned to be allocated for this research in 1995–1996[5].
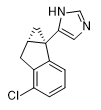
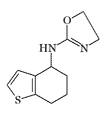
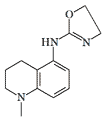
Naphthylmedetomidine (4-NEMD, YA-II-085). In 1989, at the annual chemical defense conference, a group of scientists from Edgewood Center (ERDEC) and Ohio University announced the creation of a new adrenomimetic not inferior to medetomidine in strength — naphthylmedetomidine[45]. Two years later, at the next conference, they presented results of animal experiments showing that the new substance acts more strongly than medetomidine as a sedative but has less effect on blood pressure[21]. In 1992, a patent was obtained for using naphthylmedetomidine as a sedative and hypotensive drug[24]. The (+)(S)-isomer of naphthylmedetomidine may be of even greater interest — it is much more active at α₂-receptors than its (-)(R)-counterpart[3,12].
Research on Medetomidine-type Incapacitants in Other Countries
Of particular interest for creating non-lethal weapons are cocktails of dexmedetomidine with analgesics, anesthetics, and sedatives. Surprisingly, the dose of the latter can be reduced to 5% of the usual while maintaining the same effectiveness[1]. In medicine, the most popular combination became medetomidine with ketamine — requiring very little ketamine. Moreover, medetomidine reduces ketamine's harmful effects on the nervous and cardiovascular systems.
Czech Republic. In 2001, United States Army Soldier and Biological Chemical Command (USASBCCOM) tested a triple composition on animals — ketamine, midazolam, and medetomidine — as a potential non-lethal weapon[43]. The same mixture was experimented with from 2002 at the Czech Military Medical Academy[39], with nurses volunteering as test subjects. After intramuscular injection, complete immobilization occurred in just 2–4 minutes[55].
Czech non-lethal weapons specialists studied naphthylmedetomidine from 2008, conducting experiments on mice (2008)[17], rabbits (2009)[15], rhesus macaques (2009)[18], chimpanzees and orangutans (2010)[44]. Unlike medetomidine, naphthylmedetomidine did not cause suppression of consciousness or immobilization, but only moderate sedation. But most remarkably — it completely suppressed aggression, test animals did not react to touch or any manipulations while remaining awake. The effect occurred very quickly — aggressiveness disappeared literally within 2 minutes after injection[18]. Its side effects on the cardiovascular and respiratory systems were less pronounced. Czech scientists also tested a mixture of naphthylmedetomidine, ketamine, and midazolam on primates, adding the enzyme hyaluronidase to accelerate onset[44].
Effects of naphthylmedetomidine/ketamine/hyaluronidase combination on primates
 |
 |
 |
Left — "typical" posture of rhesus macaque 5 minutes after injection (Votava, 2009), center — preserved grasping reflex against deep sedation in chimpanzee (Hess, 2010), right — immobilized orangutan (Hess, 2010)
Iran. Research on medetomidine group incapacitants is actively conducted at Iran's Imam Hussein University. In 2011, they created an aerosol composition for temporarily disabling the enemy based on a 5.5% solution of medetomidine in alcohol[46]. In 2013, the Iranians tested "optimized drug formulation with highest possible concentration of drugs": medetomidine, anesthetics ketamine and sevoflurane[47]. In 2014–2015, chemists at Imam Hussein University developed improved methods for synthesizing medetomidine and naphthylmedetomidine[52,53].
In 2014, Imam Hossein University sought kilogram quantities of medetomidine (over 10,000 effective doses) from Chinese exporters.[64] In 2023, a group opposing Iranian state-backed hackers posted leaked documents revealing an Iranian military university’s development and testing of grenades dispersing the medetomidine.[65]
People’s Republic of China (PRC) The PRC has conducted research on the synthesis of sedative adrenomimetics with enhanced potency and rapid onset, potentially suitable for use as incapacitants. The Academy of Military Medical Sciences (AMMS) synthesized medetomidine analogs that demonstrated 2–2.5 times greater potency than the parent compound. These analogs significantly inhibited spontaneous activity, exhibited potent sedative effects, and demonstrated a shorter onset time (5 minutes) with an extended duration of action (exceeding 40 minutes). Among the cited works are publications authored by American researchers from the U.S. Army Edgewood Research, Development and Engineering Center (ERDEC).[66A,C]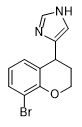 |
 |
| X2.5 medetomidine[66A] | X2 medetomidine[66C] |
AMMS also investigated combinations of medetomidine with I1-imidazoline receptor agonists, which accelerated anesthetic onset time by 40%.[66D] Additionally, they developed an effective antidote for countering dexmedetomidine toxicity based on an α1-adrenoceptor antagonist combined with a calcium channel blocker.[66B]
In 2021, the PRC, in conjunction with Iran, Russia, and Syria, opposed the Organisation for the Prohibition of Chemical Weapons (OPCW) prohibition on the law enforcement use of aerosolized central nervous system-acting chemicals. This opposition targeted proposed restrictions on α2-adrenomimetics (dexmedetomidine, clonidine) and inhaled anesthetics (halothane, isoflurane, and sevoflurane), in addition to fentanyl derivatives.[69]
The Academy of Military Medical Sciences (AMMS) and its eleven research institutes have been subject to U.S. sanctions since 2021 for utilizing "biotechnology processes to support Chinese military end uses and end users, to include purported braincontrol weaponry."[68]
Russian Federation. It appears that there is also interest in incapacitants of this group in Russia. For example, chemists from the State Research Institute of Organic Chemistry and Technology (GosNIIOKhT), that has been under US and EU sanctions since 2020 for its involvement in the development of CBW, created a new method for obtaining medetomidine that proved more effective than all previously known methods[30].
Treatment of poisoning
Antidotal therapy. For Clonidine poisoning in humans, a specific antidote is used — Tolazoline: 10 mg intravenously or 50 mg orally neutralizes the action of 0.6 mg Clonidine.
Symptomatic therapy includes infusion therapy, forced diuresis, administration of vasoactive drugs. In severe cases, extracorporeal detoxification methods are used, such as blood replacement operations and hemosorbtion (30–100% Circulating Blood Volume)[6]. Atropine eliminates hypotension and bradycardia caused by Clonidine but is rarely used for poisoning treatment. Naloxone use is also not recommended[9].
For medetomidine overdose in animals, the antidote Atipamezole (Antisedan®) is used at a dose 3–6 times the amount of medetomidine administered. The drug's action ceases within 5–10 minutes.
For non-lethal weapons, a combination of α₂-adrenomimetic with a peripheral α₂-adrenoreceptor blocker, such as Vatinoxan, is of interest, which eliminates unwanted side effects on the heart and blood vessels while preserving sedative action. Curiously, Atipamezole cannot completely block naphthylmedetomidine's action — even after its administration, animals remain indifferent and non-aggressive[44].
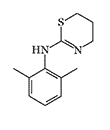
Xylazine |
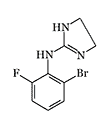
Romifidine |
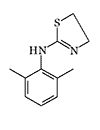
Jing Song Ling |
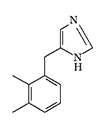
Detomidine |
M. J. Hoyer (2006) described how he accidentally injected himself with 3 mg of Romifidine (Romifidine, Sedivet®). Soon after, he experienced severe drowsiness, impaired motor coordination, and had difficulty speaking. Hallucinations occurred, his speech became incoherent, and he lost consciousness. When he was brought to the hospital, his blood pressure had dropped to 70/50 mmHg and his pulse rate decreased to 30 beats per minute. After symptomatic treatment, his condition improved[8].
| Formula | Effective Concentration IC (mg·)min/m3 |
Dosage (ID) mg/kg |
LD50 mg/kg |
Therapeutic Index |
|---|---|---|---|---|
|
1200–2000 +2400–4000 scopolamine (monkeys)[20/13] |
— | 56 (mice, iv) |
— |
 |
1200–2000 +2400–4000 scopolamine (monkeys)[20/13] |
— | — | — |
 |
3000 (gerbils)[20/8] |
2 (rats, ip) |
— | more than 30 |
 |
3000 (gerbils)[20/10] |
— | — | more than 30 |
|
2500 (monkeys)[20/9,10] |
— | 42 (mice, iv) |
more than 50 |
|
5000 (mice)[20/11] |
0.02 (mice, iv) |
4.2 (mice, iv) |
250 |
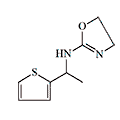 |
2000 (mice)[20/11] |
0.01 (mice, iv) |
— | 1300 |
|
— | 0.032 (mice, iv) |
20 (mice, iv) |
625[19] |
| — | 0.0056 (rats, iv) |
22.4 (rats, iv) |
400[19] | |
| — | 0.01 (dogs, iv) |
1.5 (dogs, iv) |
150[19] | |
|
— | 0.01 (mice, iv) |
22.4 (mice, iv) |
2240[19] |
| — | 0.0018 (rats, iv) |
3.16 (rats, iv) |
1755[19] | |
| — | 0.001 (dogs, iv) |
1.5 (dogs, iv) |
1500[19] | |
|
— | 0.01 (mice, iv) |
31.6 (mice, iv) |
3160[19] |
| — | 0.018 (rats, iv) |
56.2 (rats, iv) |
3120[19] | |
| — | 0.1 (dogs, iv) |
>5 (dogs, iv) |
50[19] |
Currently, alpha-2 adrenomimetics are still considered the most promising among chemical incapacitants, along with benzodiazepines and neuroleptics, which are considered to a lesser extent. However, the scope of achievements in this field remains unknown due to the classified nature of such research.