Incapacitating Agents Causing Temporary Paralysis

The impala shows no slightest signs of fear or aggression after the Quiloflex injection (Van Niekerk et al., 1963)[25]
During the 20th century, researchers explored chemical compounds that could temporarily paralyze without causing lasting harm. These agents — ranging from plant alkaloids like coniine to synthetic muscle relaxants — were studied for potential use in non-lethal warfare and animal immobilization.
Though some showed promise in early trials, most were ultimately rejected due to high toxicity, inconsistent effects, or ethical concerns. This article reviews the most notable candidates and the scientific context behind their development.
Alkylchloracetylenes (Alk–C≡C–Cl). In 1938, German chemist W. Bossaller conducted doctoral research on the synthesis of lower aliphatic alkylchloroacetylenes. During the course of this investigation, he experienced severe and prolonged disturbances of the motor nervous system, manifesting as weakness in the lower extremities persisting for several weeks, which temporarily rendered ambulation impossible. Additionally, transient signs of optic nerve paralysis were documented.[41]
By 1944, German intelligence services acquired information suggesting that Soviet researchers were investigating these compounds as potential chemical warfare agents. This research direction in the USSR was apparently initiated due to an erroneous assumption that parallel work was being conducted in Germany.[42]
Nicotine and Coniine. After World War I, British authorities considered nicotine as a potential chemical warfare agent. However, they ultimately abandoned this idea due to nicotine's lack of clear advantages over already established chemical weapons.[2]
During the 1950s and 1960s, nicotine was tested in dart guns as an immobilizing agent for wild animals. Unfortunately, the extremely high mortality rate among paralyzed animals forced veterinarians to switch to safer alternatives.[3]
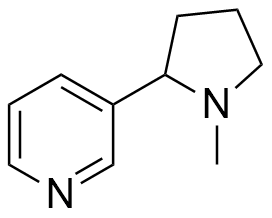
Nicotine |
Coniine |
γ-Coniceine |
Coniine, the toxic alkaloid found in poison hemlock (Conium maculatum), produces effects similar to nicotine, but with more pronounced paralytic properties.[38] This compound ranks among history's oldest sabotage poisons — during the Venetian–Florentine War (1467–1468), the Florentines used hemlock to contaminate the water in a canal near the enemy camp. To this end, the commander of the Republic of Florence’s forces
What makes coniine particularly dangerous is its volatility at room temperature. Even inhaling small amounts of its vapors can trigger dizziness, nausea, and headaches in humans.[35] At higher doses, coniine causes ascending paralysis.
During World War II, Coniine and γ-Coniceine were synthesized at the University of Wisconsin[43] and considered as potential chemical warfare agents, but were found to be unsuitable.[31]
Central muscle relaxants. In 1959, for the US Congressional committee inquiring into CBW, a demonstration was conducted showing the effects on animals of two incapacitating agents — CS 4640 (Etonitazene) and an unknown muscle relaxant. About the latter, it is only known that its calculated effective concentration for humans through inhalation exposure was approximately 1000 mg·min/m³, and its effects lasted from ½ to 2 hours or longer, after which spontaneous recovery occurred. As stated, this substance was actively tested on volunteers.[17]
In 1960, the Director of Medical Research at the U.S. Army Chemical Warfare Laboratories, speaking at a conference of military surgeons, described a new paralyzing chemical agent:
The chemical structures of the agents described above remain classified to this day, but the reported clinical presentation is characteristic of poisoning with substances from the group of centrally acting muscle relaxants. Here is how their effects are described in the Soviet manual Toxicology of Psychic- and Physical- Incapacitants (1970):
Central acting muscle relaxants are limited in medical application for treating spastic conditions of various origins. All known pharmacological agents in this group are unsuitable as incapacitating agents, as they induce paralysis only at dosages close to lethal levels. The most well-known muscle relaxant in this group — Mephenesin, causes only mild muscle weakness at intravenous doses of 700–1800 mg, without any narcotic effect.[59] Fatal cases have been reported following intravenous administration of 3900 mg[60] and oral intake of 7500 mg.[20]
1,4-Benzodioxanes. In the early 1960s, myorelaxants of the 1,4-benzodioxane group demonstrated the greatest potential as incapacitating agents. At the British Chemical Defence Experimental Establishment (CDEE), Ethomoxane was considered comparable to the most potent analgesics of that period — etonitazene and phenylpiperidinols.[36] Regarding sedative effects, Ethomoxane and its chloro-analog proved 20 times more potent than chlorpromazine, one of the strongest central nervous system depressants of the 1950s. Information about this substance's effects on humans remains limited; however, mice administered this compound exhibited a notable reduction in their aversion to handling.[36] 5-Cloroethomoxane[45] underwent toxicological testing in the chemical laboratory of Edgewood Arsenal.[46]
The limited published literature confirms CDEE researchers' collaboration with pharmaceutical companies in studying compounds from this group,[22] as well as myorelaxants derived from dimethylphenethylurea,[23] tetrahydropyrimidines,[24] and other chemical classes.
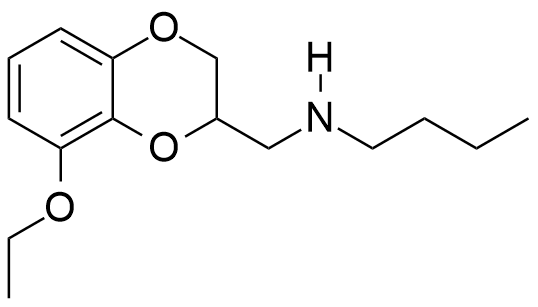
In general, muscle relaxant effects are not characteristic of 1,4-benzodioxane derivatives and have been documented in only two pharmaceutical agents — Quiloflex and Ambenoxan.
 |
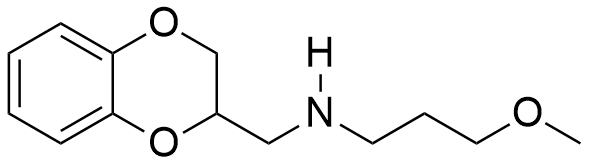 |
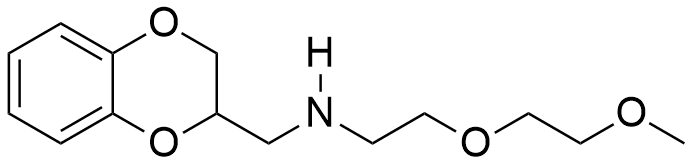 |
| Ethomoxane | Quiloflex | Ambenoxan |
Synthesized in the late 1950s, Quiloflex failed to find applications in human medicine due to its frequent induction of nausea, emesis, and vertigo in patients. In 1962, veterinarians developed interest in the compound, which subsequently demonstrated promising results in the chemical immobilization of antelopes and giraffes. Within 1–2 minutes post-injection, the animals exhibited ataxia, decreased mobility, reduced responsiveness, and a tendency to assume recumbent positions. Quiloflex did not produce pronounced hypnotic effects; however, even those specimens that maintained standing posture displayed no indications of aggressive behavior.[25] Notably, canine subjects manifested markedly different responses to Quiloflex administration — domesticated dogs previously exhibiting prosocial behavior toward humans demonstrated fear responses post-injection, retreating to corners and becoming unresponsive to any attempts at coaxing them from these locations.[26] Unusual effects have also been described for the thiol analog of quiloflex — it induces aggressive behavior in male rats 12 to 24 hours after administration.[44]
Ambenoxan at a dosage of 5 mg/kg induced skeletal muscle relaxation in primates, without demonstrating a pronounced sedative effect. Similar to quiloflexe, it does not cause complete paralysis, but rather produces muscle weakness.[28] Both compounds are unsuitable as incapacitating agents due to their low activity and high toxicity.
Oximes. Discovered in the mid-1950s, dicyclopropylketoxime is a rapidly acting compound that produced flaccid paresis in dogs and monkeys.[26] Its potency, however, was no greater than that of mephenesin. Between 1962 and 1964, Colgate Palmolive Co. collaborated with the U.S. Army Chemical Research, Development and Engineering Center (CRDEC) on a program to identify new incapacitating agents, including oximes structurally related to the centrally acting muscle relaxant cyclobenzaprine.[27] Oximes of benzocyclobutene and 1,4-benzodioxane were likely part of the study.[47] Some of these compounds proved to be especially effective muscle relaxants — more potent than mephenesin and significantly safer. The lethal doses for mice exceeded 3200 mg/kg per day,[47] which is several times less toxic than ordinary table salt.[48]
 |
 |
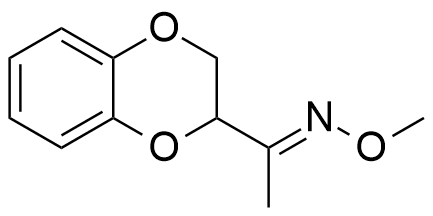 |
| Compound Colgate Palmolive Co | ||
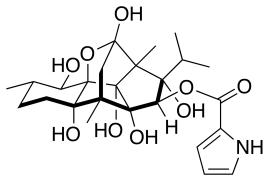
Ryanodine From 1958 to 1961, the U.S. Army Chemical Corps continued its investigation of ryanodine and established several contracts with the University of California, with the objective "to isolate in pure form and to determine the chemical structure of ryanodine which has an unusual type of pharmacological activity of interest in the search for agents".[14] Research activities were subsequently continued at the Edgewood Arsenal beginning in the early 1960s.[16] One of the participants in the U.S. non-lethal chemical agent development program mentions another compound that induces spastic paralysis: 1-(trichloromethyl)cyclohexan-1-ol.[37]
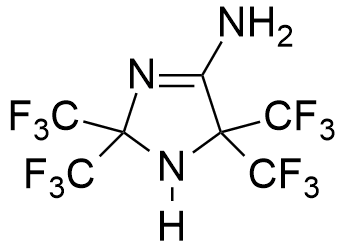
Midaflur. In the mid-1960s, it briefly seemed that scientists had finally solved the challenge of creating a safe yet powerful central muscle relaxant. The story begins with chemist William J. Middleton of DuPont, who discovered crystalline residue at the bottom of a flask following one of his experiments. After further purification, Middleton and a colleague decided to submit this unknown substance for biological testing.[54]
The results were astonishing — the new compound, later named Midaflur, proved to be the most potent central muscle relaxant known at that time,[49] while also functioning as a relatively safe anesthetic. In one remarkable experiment, dogs injected with Midaflur fell into a deep sleep lasting an entire week. They briefly awoke only to drink water before returning to sleep for another two days.
Surprisingly, this prolonged hibernation-like state had no apparent negative effects on the animals' health. However, since the dogs received no food during their extended slumber, they lost considerable weight by the experiment's conclusion. This led the chemists to joke that they had inadvertently invented the perfect solution for obesity treatment.[54]
However, midaflur did not meet the expectations of pharmacologists, as the anesthesia was superficial and the muscle relaxation was incomplete.[50] Despite close collaboration between DuPont and the US Army in developing incapacitating agents, there is no documentary evidence of military interest in midaflur.
 |
 |
|
| Ryanodine | Midaflur | Benzothiazioles |
In 1967, prominent chemical weapons expert J.P. Robinson noted potential incapacitating agents causing temporary paralysis, specifically benzothiazole derivatives.[29] Initial investigations of this myorelaxant group commenced in 1952. The most active aminobenzothiazole derivatives, at a dosage of 15 mg/kg, induced flaccid paralysis, ataxia, emesis, hypersalivation, and frequent micturition in experimental animals. The primary limitation of aminobenzothiazoles was their high toxicity — the lethal dose was only marginally higher than the paralyzing dose.[30]
The development of incapacitating agents with paralyzing effects on humans was also carried out in other Western Bloc countries. In 1980, a report from West Germany mentioned the development of a "riot gas which would immobilize demonstrators for up to half an hour without causing harmful side-effects or making them unconscious".[34]